Virtual Screening Workflow
Introduction
Virtual screening (VS) is a computational technique in drug discovery that identifies potential drug candidates from compound libraries. It uses computer-based methods to prioritize compounds for further testing based on their predicted interaction with specific biological targets, like protein receptors or enzymes. VS efficiently narrows down the search for new drug candidates, saving time and resources by focusing on the most promising compounds. It's a vital step in drug discovery, helping identify lead compounds for further therapeutic development
Hermite's Virtual Screening Workflow module offers high-throughput virtual screening capabilities. It offers ligand property-based filtering tools, 4 restrictive docking methods (H-Bond, Maximum Common Structure, Substructure, Shape), and integrates models like Uni-Clip (rapid ligand screening via a contrastive learning algorithm), Uni-Dock, Uni-Mol (pre-training model for optimizing docking poses) to significantly improve screening speed and accuracy. Additionally, Hermite boasts an extensive built-in virtual compound library (22 million), enabling swift virtual screening across expansive chemical spaces.
Sigma 2 Receptor:
The sigma 2 receptor is often overexpressed in proliferating cells and many tumors. The marked sigma 2 receptor is proposed as a tool for cancer diagnosis and treatment and is also considered a target for Central Nervous System diseases. This tutorial focuses on virtual screening molecules against the sigma 2 receptor, specifically using 7M94, a sigma 2 receptor ligand in conjunction with Roluperidone (a sigma 2 receptor antagonist) [2], as a reference for screening within our database.
The following files are used in this tutorial:
1. Virtual Screening Workflow
1.1 Function Entry
Access the Virtual Screening Workflow by navigating to the left common menu bar: Function → Virtual Screening → Virtual Screening Workflow.
Click Yes in the "MM PB/GBSA" pop-up window.
1.2 Selecting a Prepared Protein
Click Select File and upload the local file "7M94_prep.pdb".
The uploaded protein will undergo force field inspection automatically. Once the status changes from "Processing" to "Valid," click Next.
Note: If the force field test fails, use Protein Preparation in the upper right corner to re-validate. For detailed instructions, refer to "Protein Selection, Ligand Selection and Pocket Setting".
1.3 Ligand Preparation
1.3.1 Selecting Prepared Ligands
Click Select from Database. A "Select Ligands" window will appear; choose the dataset "Specs," then click OK.
Click Next.
Note: Prior to virtual screening, ligands must undergo preparation, including isomer generation, hydrogenation, protonation, energy minimization, etc. Ligands in Hermite's database have already been processed, so the ligand preparation will be skipped. For detailed instructions on ligand selection methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
1.3.2 Filters Setting
You have the option to filter the ligands according to their properties (e.g. Molecular weight, number of Hydrogen Bond donors, etc).
1.4 Pocket Setting
Click the Select File button to upload "pocket.txt".
Note: For detailed instructions of pocket setting methods, see "Protein Selection, Ligand Selection and Pocket Setting".
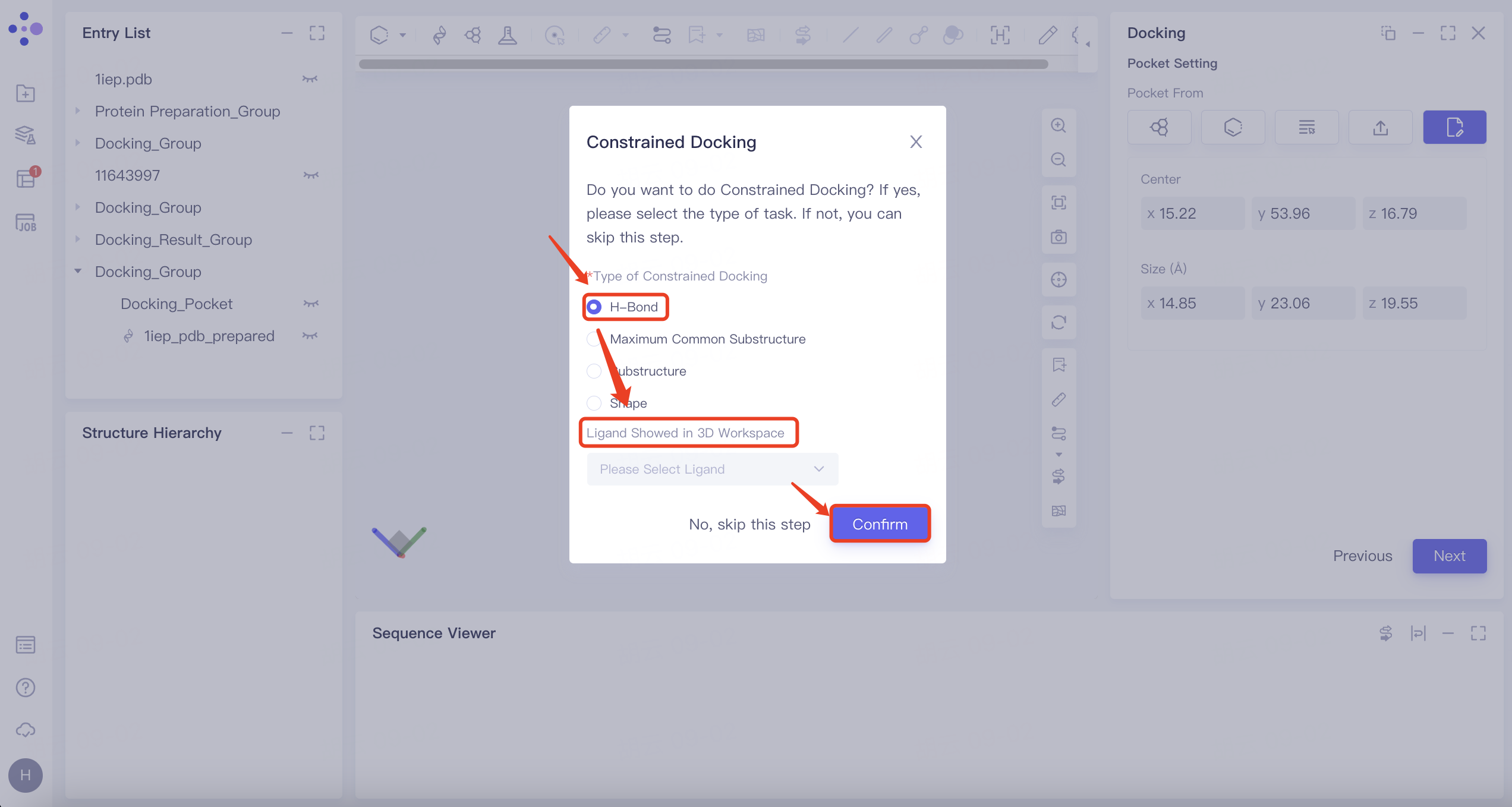
1.5 Constrained Docking
Select No, skip this step here to skip this step.
Note: Constrained docking is necessary in some cases. Refer to the table below for more information:
| Constrained Docking Type | Instruction | Display | Note |
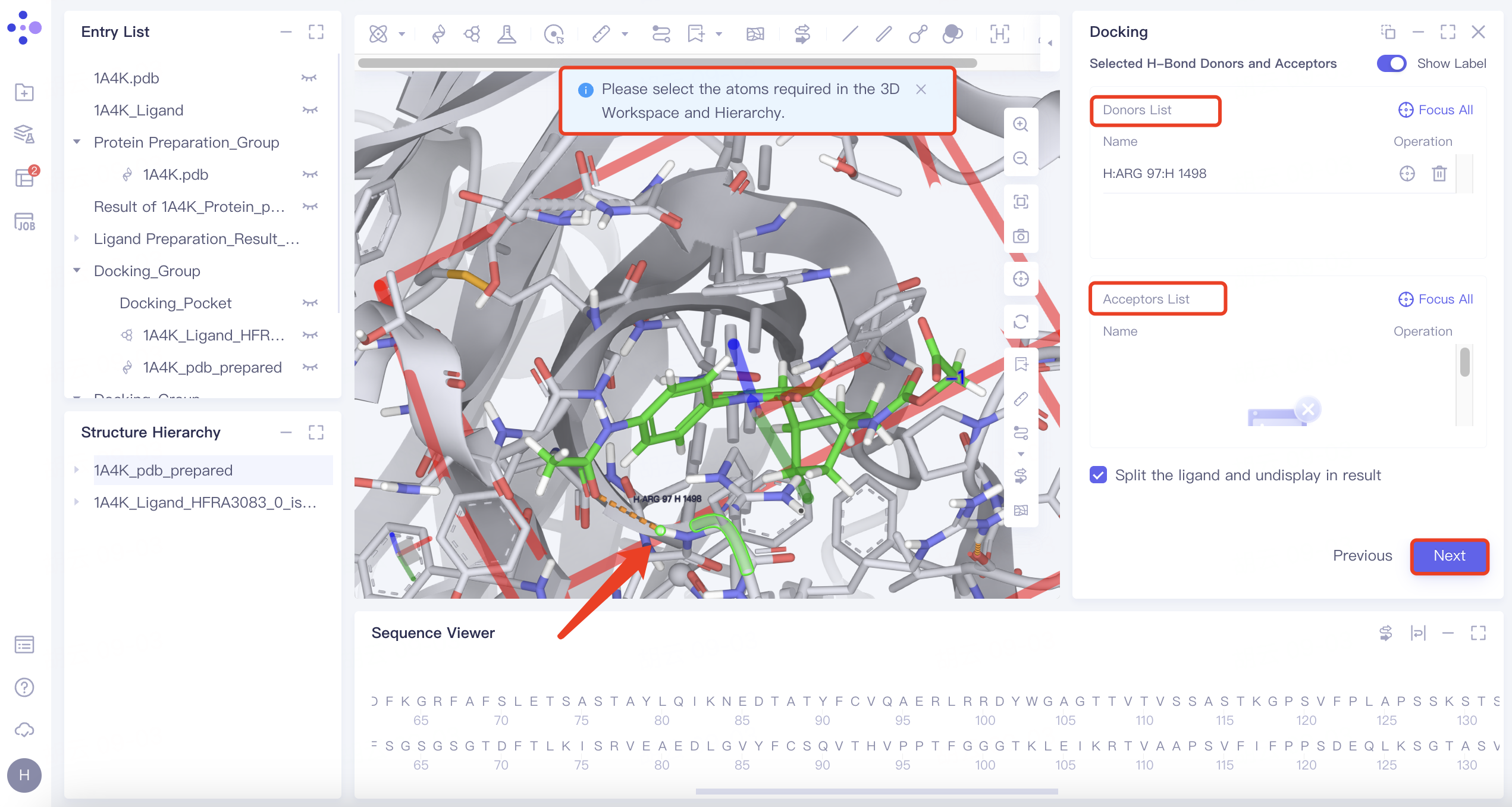
| H-Bond | 1) Select "H-Bond" and choose the reference ligand displayed in "Ligand Showed in 3D Workspace". Click "Confirm". 2) In the 3D Workspace window, select atoms in the acceptor that interact via hydrogen bonds with the ligand. These atoms will be automatically categorized into the hydrogen bond acceptor and donor list on the right. |
| 1) When certain hydrogen bonds formed between ligands and acceptors are associated with molecular activity, new molecules with potential activity can be screened by H-bond constrained docking. 2) The orange dashed line indicates the hydrogen bond interaction that currently exists between the ligand and the acceptor. |
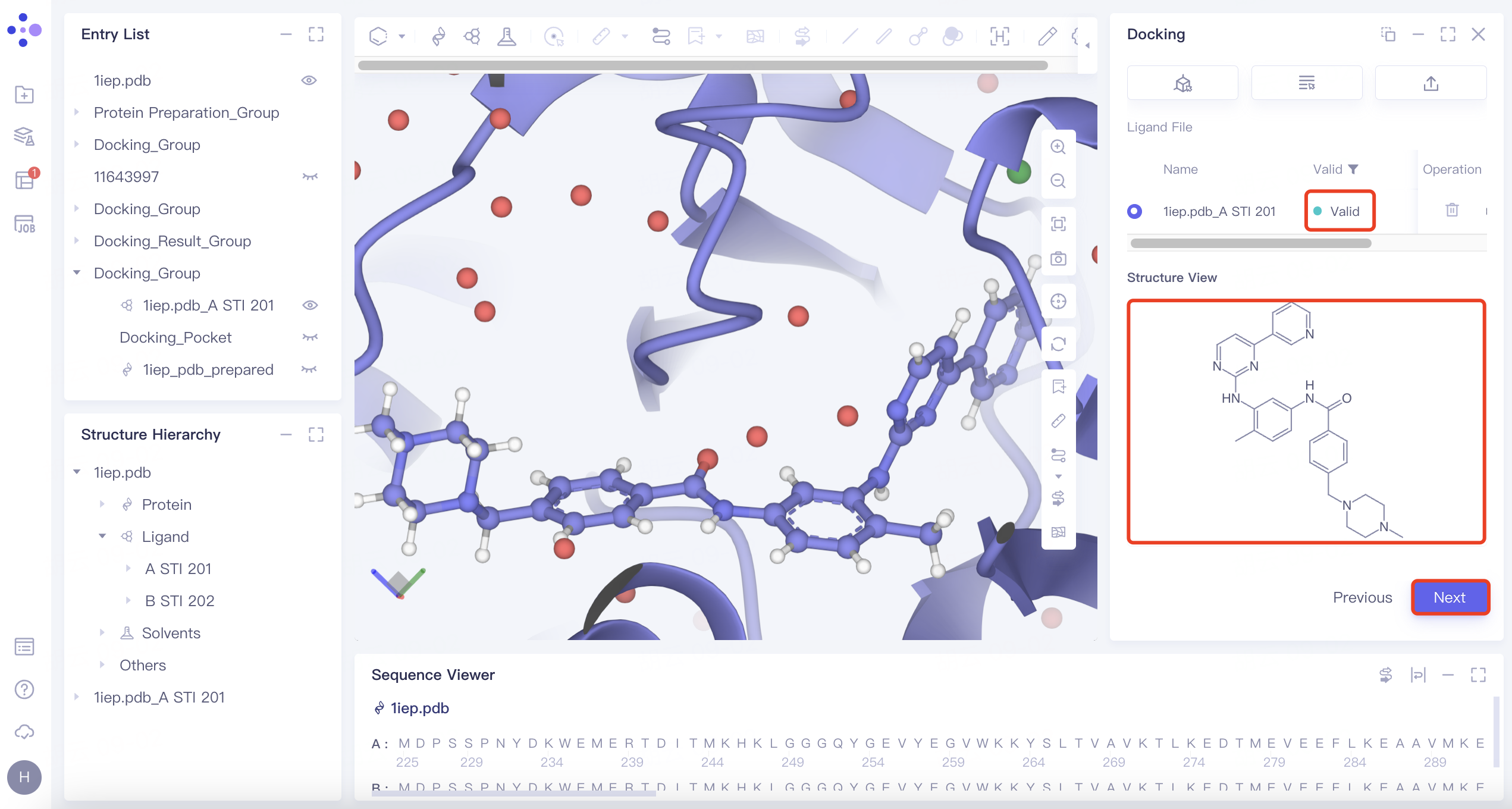
| Maximum Common Substructure | 1) Click the "Maximum Common Substructure" button and then click "Confirm". 2) Select the reference ligand (the ligand with the desired binding pattern to the receptor). The ligand will undergo force field inspection, and upon passing, the "Valid" status will be displayed in the Valid column. Simultaneously, the 2D molecular diagram of the ligand will appear in the "Structure View" window. |
| Use "Maximum Common Substructure" when you have identified a ligand with an effective binding pose to a receptor and wish to identify other molecules that share this binding pose. |
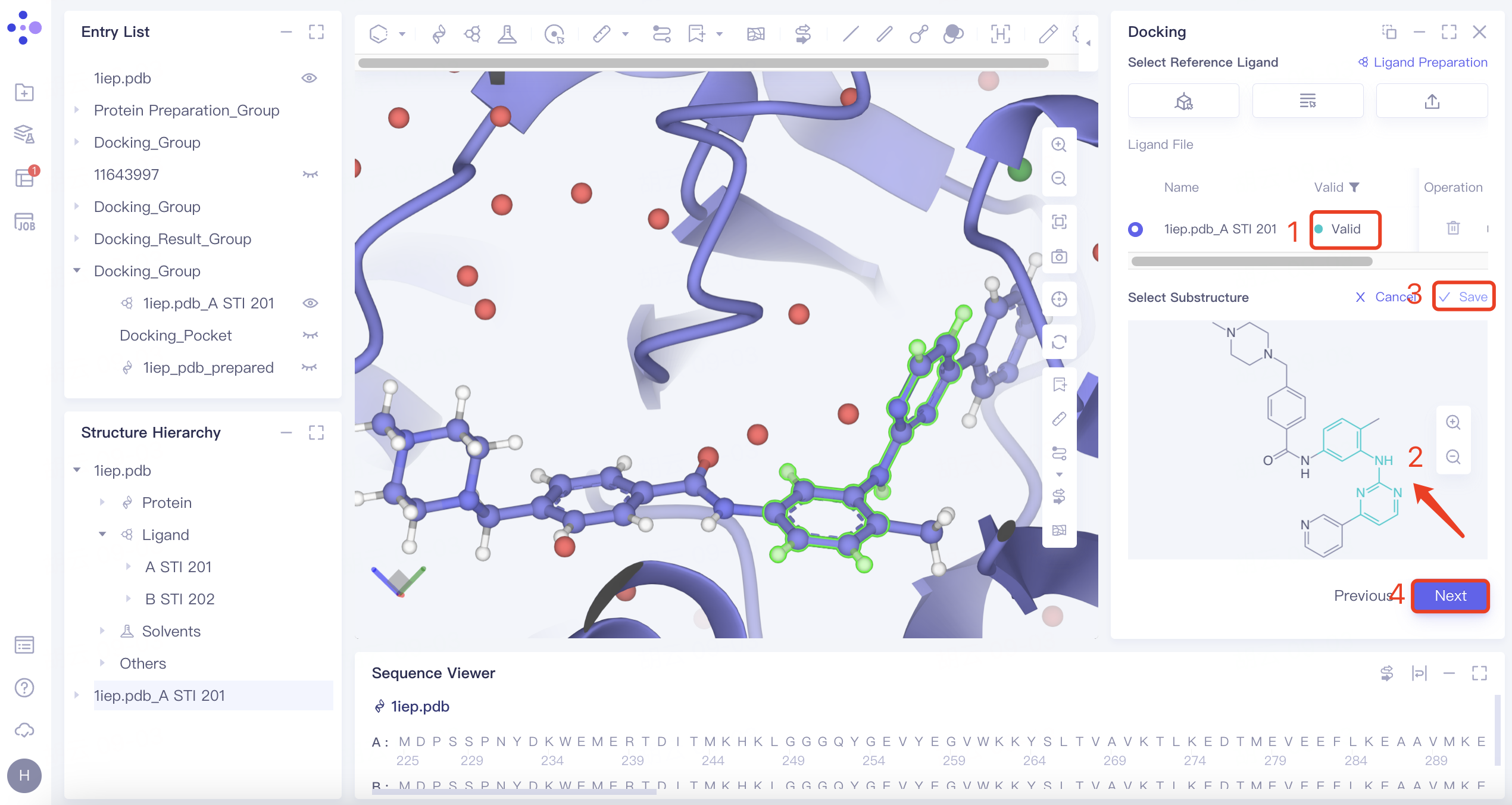
| Substructure | 1) Click the "Substructure" button, then click "Confirm". 2) Select the reference ligand (a ligand with specific substructures). Upload the ligand for force field inspection. Once it passes the inspection, "Valid" will be displayed in the Valid column. The 2D molecular diagram of the ligand will appear in the Structure View window. After selecting the substructure, click "Save" to store the substructure information. |
| The Substructure option facilitates molecular docking by focusing on substructure screening. This approach is used to search for ligands that contain specific substructures, aiding in targeted docking processes. |
| Shape | 1) Click the "Shape" button, then click "Confirm". 2) Select the reference ligand (a ligand with a specific shape). Upload the ligand for force field inspection. Once it passes the inspection, "Valid" will be displayed in the Valid column. Define the shape by expanding the van der Waals radius of the reference ligand atoms. Click "Sync to 3D" on the right to visualize the shape in the 3D Workspace window. |
| 1) Shape-based virtual screening is crucial for skeleton transition, biological isomer substitution, and virtual library design. The "Shape" option enables shape-based virtual screening. 2) or example, setting the ratio to 20% means expanding the van der Waals radius of the original molecule by 20% to create a new volume. |
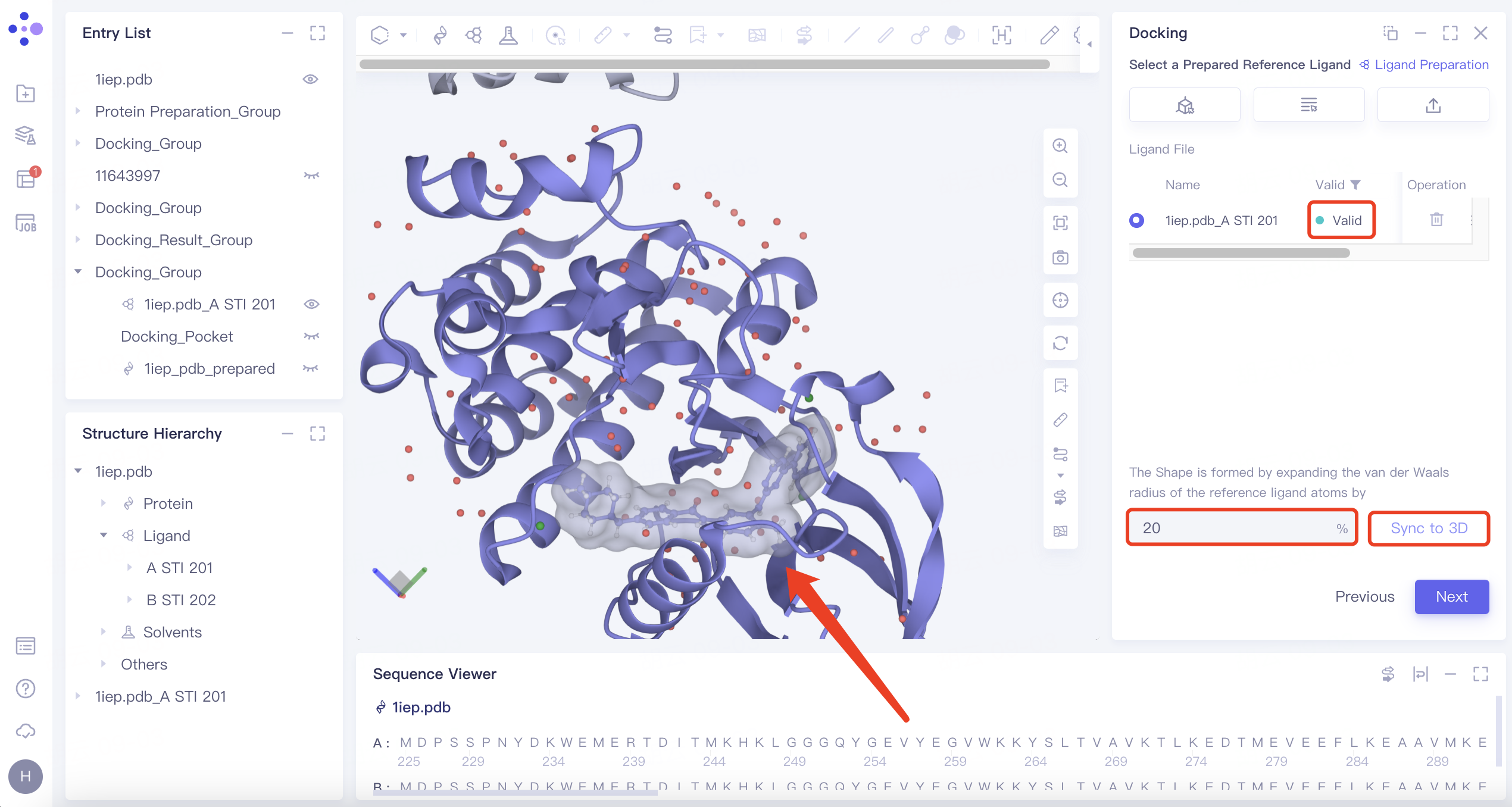
1.6 Workflow Setting
Check Use Uni-Clip to retain ligands with the top 50% of rankings and adjust 50% to 70%;
Check 1st: Fast Mode Docking
Set Number of Results to Keep to 1st Docking to top 20%
Leave all other parameters at default values;
Check 2nd: Balance Mode Docking
Set Number of Results to Keep to 1st Docking to top 20%
Leave all other parameters at default values;
Check 3rd: Detail Mode Docking
Set Number of Results to Keep to 1st Docking to top 20%
Leave all other parameters at default values;
Check Use Uni-Mol to optimize the resulting ligands' pose(s) to optimize the docking conformation;
Check Rescore, and select Gnina as the scoring function;
For MM PB/GBSA Settings, keep all parameters at default values;
Select Next, and keep the default Job Name;
Click Submit.
| Options | Explanation | Note |
| Uni-Clip | A tool based on a comparative learning algorithm that quickly filters out ligands with unfavored conformation | It is a crude screening method, and it is recommended to retain 70% of the results |
| Fast/Balance/Detail Mode Docking | Fast:exhaustiveness 128 & max_step 20 | Three different search modes entail different levels of complexity for molecular docking calculations. The 'Detail' mode offers the most comprehensive coverage of the search space and has the highest accuracy. This is followed by the 'Balance' mode, with 'Fast' being the least comprehensive and accurate. |
| Balanced:exhaustiveness 384 & max_step 40 | ||
| Detailed:exhaustiveness 512 & max_step 40 | ||
| Number of Results to Keep to 1st/2nd/3rd Docking | Can choose to keep All/Top x/Top % results | —— |
| Keep Multi Binding Poses for Each Ligand | 1) Number of Binding Pose: supports generation of up to 100 conformations 2) Energy Range (kcal/mol): The maximum energy difference allowed from the optimal binding mode | —— |
| Scoring Function | Supports Vina, Vinardo, AutoDock4 | Vina is generally preferred |
| Uni-Mol | Optimize the pose after docking using Uni-Mol | Essentially a Constrained Docking based on Machine Learning |
| Rescore | Re-score the docking conformation using Vina, Vinardo, GNINA, KarmaDock Affinity, RTMScore, Uni-Mol Affinity, or Uni-Score | Gnina is generally preferred |
Note: For MM PB/GBSA parameter settings, refer to "MMPB/GBSA".
2. Result Analysis
2.1 Access the Results
Access the job results by clicking Job on the left common menu bar. The task generated four result files: "7M94_protein_prep_Clip", "7M94_protein_prep_VSW", "7M94_protein_prep_Rescore", and "7M94_protein_prep_GBSA".
Note: Virtual Screening Workflow includes up to five subtasks as follows:
| Job Type | Explanation |
| Ligand Prep | This task is generated when ligands are not imported using Hermite's provided databases, and undergo the preparation process. |
| Uni-Clip | This task is generated when "Uni-Clip" is selected during the workflow settings. |
| VSW | These are the final results after "Fast", "Balance" and "Detail" docking searches. |
| Docking Rescore | This task is generated when "Rescore" is selected. |
| MM PB/GBSA | This task is generated when "MM PB/GBSA calculation" is selected. |
2.2 Uni-Clip Results
"7M94_protein_prep_Clip" is the preliminary screening results by Uni-Clip.
Note: The download result includes a table file which contains three columns: Ligand ID, Ligand Name, and Clip Score (docking scores given by Uni-Clip, the higher the better).
2.3 VSW Result
"7M94_protein_prep_VSW" contains the docking results. Click Show in the "Operation" column to view the result table.
The docking results are sorted by "Score" by default.
Docking results table explanation:
| Column Name | Description | Note | |
| Rank | Pose ranking by scoring function. | —— | |
| Stars | Option to mark a favored pose | —— | |
| Score | The score value assigned to the pose after docking with Fast, Balance and Detail search modes | The scoring function used is the one selected for the final search mode | |
| [Constrain Type] Score | Constrained Docking score | This column is available only when Constrained Docking is selected | |
| [Rescore Function] Rescore | The value generated by the rescoring scoring function | A "(+)" sign indicates that higher scoring function values are preferable, whereas a "(-)" sign suggests that lower values are more desirable | |
| Operation | Fix | Fixed display in 3D Workspace | —— |
| 3D | Display the ligand in 3D Workspace | —— | |
| Property | View property information | —— | |
| Download | Download this pose | Data are saved in sdf format | |
To view a pose in 3D, click the 3D button on the right side of the top-ranked pose in the Rank ranking. This displays it in the 3D Workspace window. You can switch between poses using the up and down keys on your keyboard. Favored poses can be marked with a star.
The ligand is shown in a ball-and-stick model, while interacting amino acid residues are depicted as lines. Dotted lines represent the interactions. Clicking or hovering over these dotted lines will display a description of the interaction in the lower-left corner of the 3D Workspace window. Different interactions are indicated by varying colors.
Note: The "Show" function can display up to the top 3000 ligands. Full data is accessible by downloading it from the Job section.
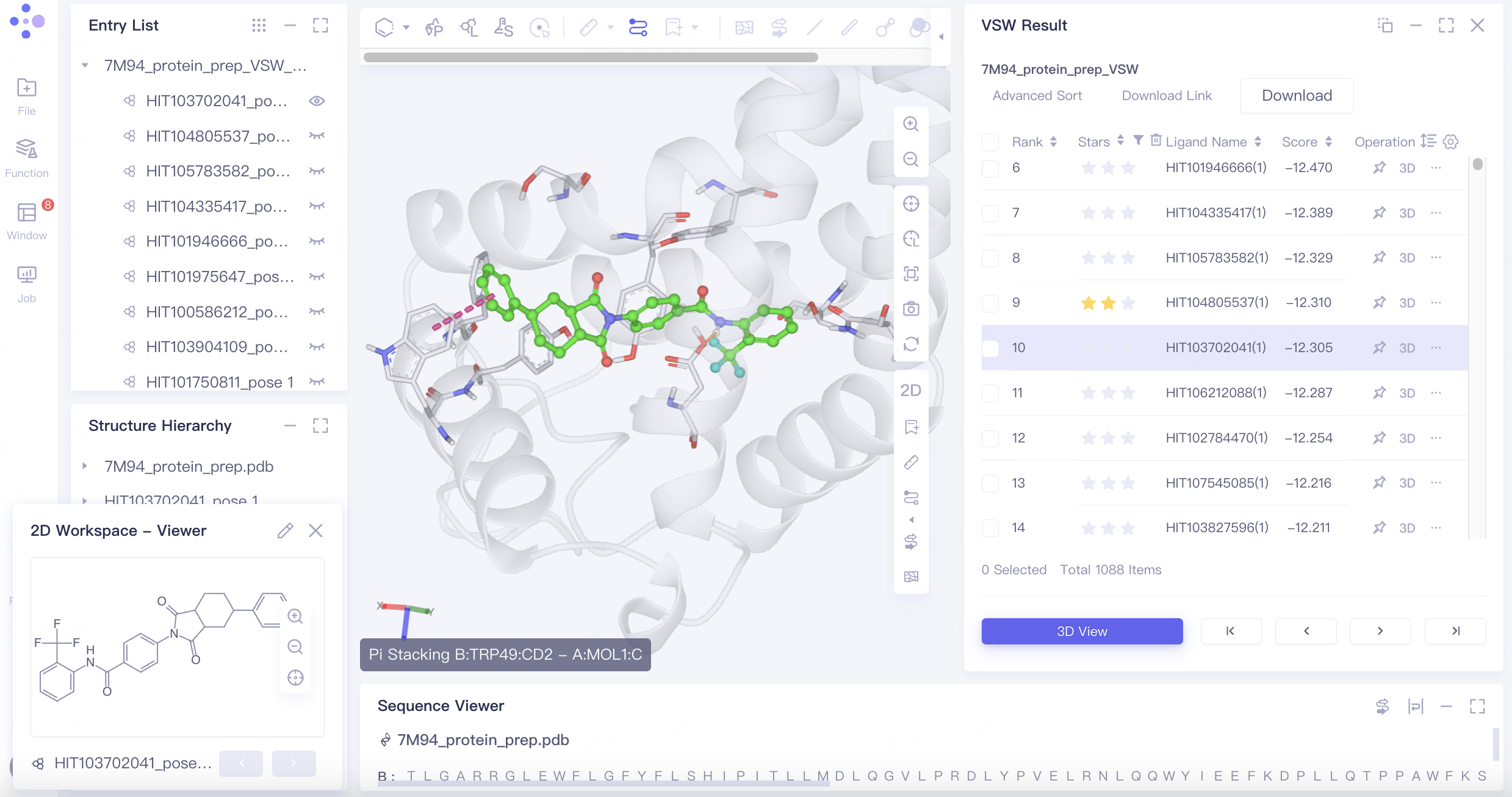
The Pi-Pi Stacking interaction between Tyr150 and the ligand may be linked to the ligand's activity. To screen for poses with this interaction, follow these steps:
Click Advanced Sort and select IFP Filter in the popup interface. Choose the options shown below in the heatmap, click Confirm, and the filtered results will be reorganized in the results table.
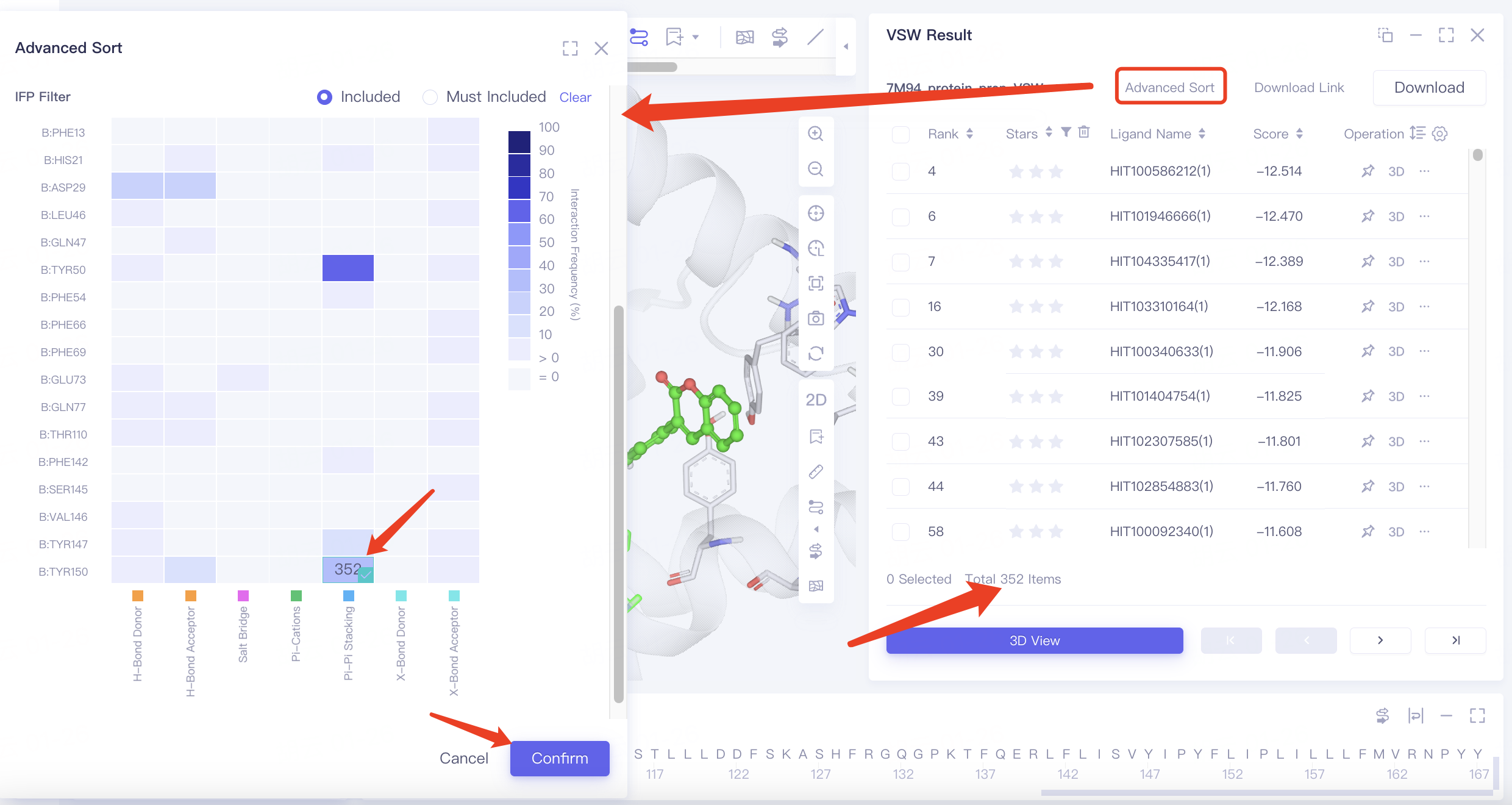
Note: Advanced Sort allows for filtering of poses using various methods:
Result Filter: Filters docking results based on the range of Star, Score, and Rescore values.
Result Rank: Sorts docking results by Ligand Name, Star, Score, and Rescore.
IFP Filter: Filters docking results based on interactions between ligands and proteins.
"Include" means that the docking result is retained if it contains any selected interaction.
"Must Include" requires that the docking result contains all selected interactions to be retained.
Heatmap explanation:
The horizontal axis represents interaction types, and the vertical axis shows the residue and its sequence number. The color intensity indicates the frequency of interactions between ligands and residues in the docking results.
Hovering over a square in the heatmap reveals the number of docking results featuring that particular interaction.
2.4 Rescore Results
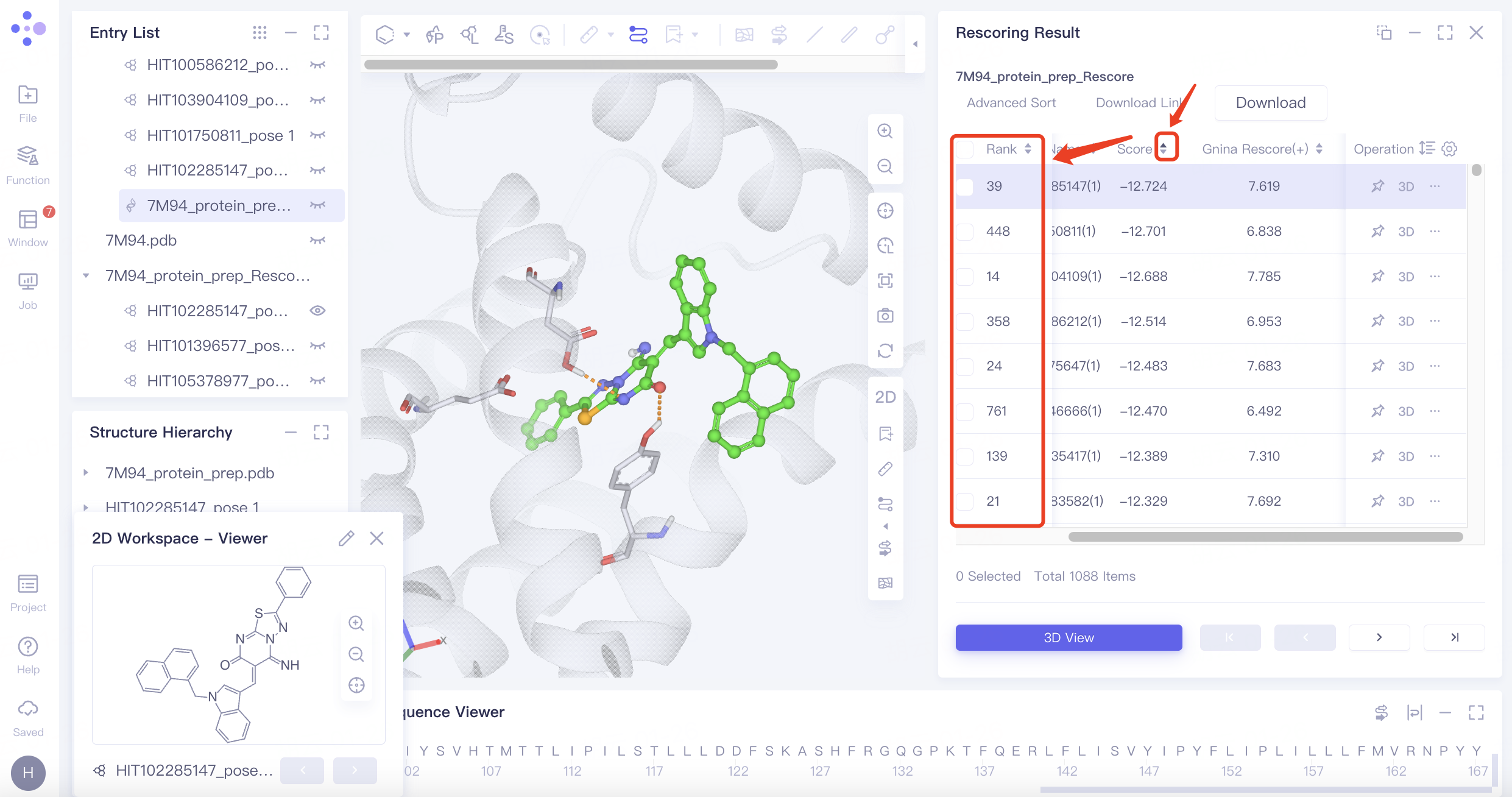
"7M94_protein_prep_Rescore" is the Rescoring result of the task. Different from "7M94_protein_prep_VSW", it sorts the docking results by the new "Gnina Rescore (+) " column scores.
Rescoring provides more scoring function options, which may lead to differences in the virtual screening results. In this tutorial case, for example, the pose with the highest score in Score (Vina) ranked only 39th using Gnina rescoring. Rescoring provides more comprehensive screen results.
2.5 MM PB/GBSA Results
MM PB/GBSA calculates the binding free energy between ligands and proteins, which offers more accurate ligand affinity ranking than most molecular docking scoring functions.
"7M94_protein_prep_GBSA" is MM PB/GBSA calculation result for this task as shown in the figure below. Click 3D under the "Operation" column on the right to display each pose involved in the 3D Workspace. Molecules can be further screened by ΔG Total and molecular conformation.
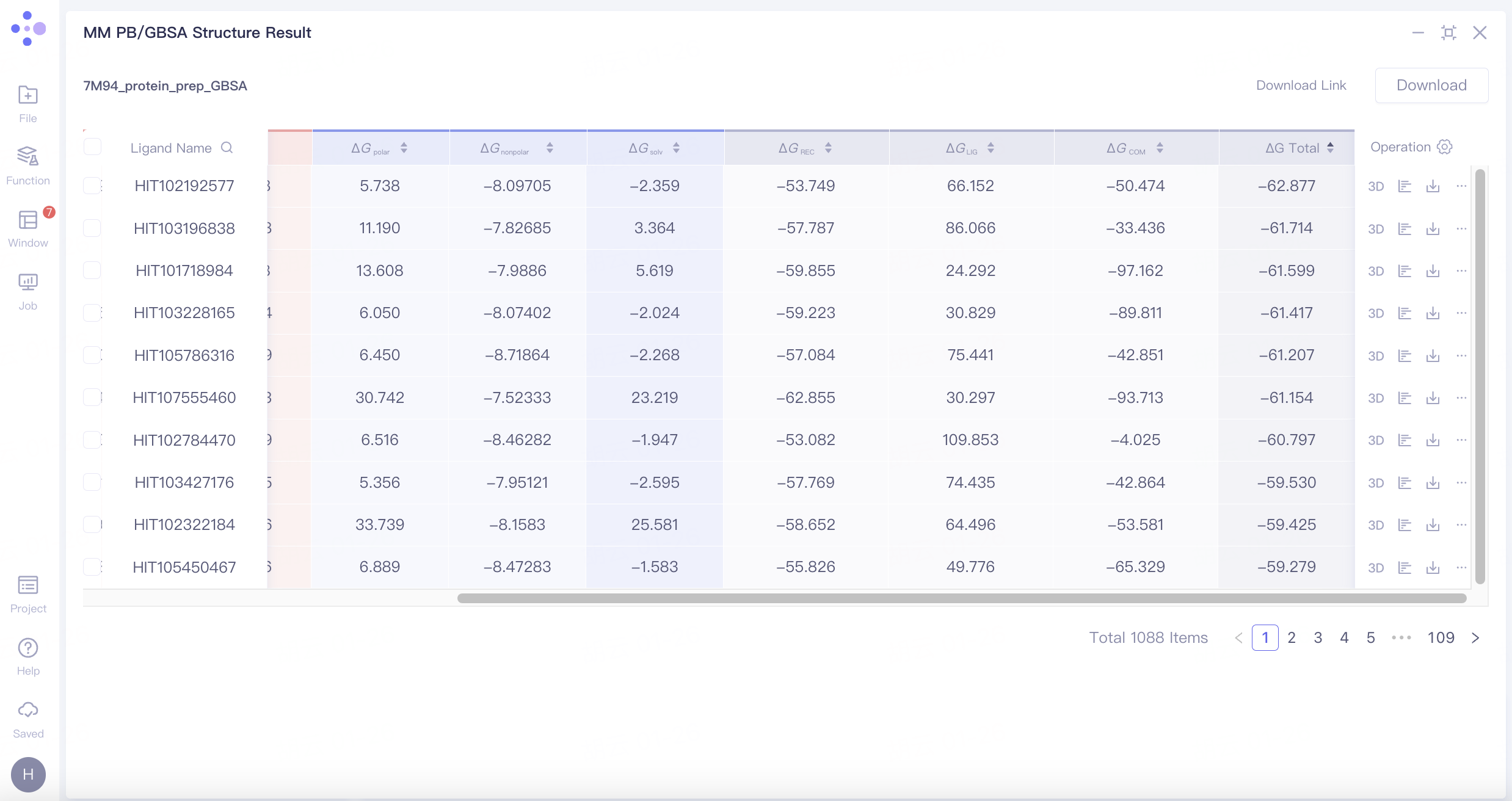
MM PB/GBSA calculation provides the contributing ΔG breakdown:
Note: See "MM PB/GBSA" for more details.
3. References
[1] Yu Y, Cai C, Wang J, et al. Uni-Dock: GPU-accelerated docking enables ultralarge virtual screening[J]. Journal of Chemical Theory and Computation, 2023, 19(11): 3336-3345.
[2] Alon A, Lyu J, Braz J M, et al. Structures of the σ2 receptor enable docking for bioactive ligand discovery [J]. Nature, 2021, 600 (7890): 759 764.